Αμυλοείδωση: εκδηλώσεις από το ήπαρ
H αμυλοείδωση δεν αποτελεί μία νόσο, αλλά τον όρο που χαρακτηρίζει ευρύ φάσμα διαταραχών με κοινό χαρακτηριστικό την εναπόθεση παθολογικών αδιάλυτων πρωτεϊνικών ινιδίων στον εξωκυττάριο χώρο οργάνων και ιστών.
Γράφει η
Έλενα Βεζαλή
Παθολόγος – Ηπατολόγος
Συνεργάτις Ηπατολογικού Τμήματος ΥΓΕΙΑ
Οι κλινικές εκδηλώσεις της αμυλοείδωσης ποικίλλουν ανάλογα με τον τύπο, την εντόπιση και την ποσότητα των εναποθέσεων. Οι πρωτεΐνες που σχηματίζουν τις αποθέσεις αμυλοειδούς, προέρχονται από διαλυτές πρόδρομες ουσίες που έχουν υποστεί δομικές αλλαγές της αναδίπλωσης και μετατρέπονται σε αδιάλυτα ινίδια.
Διακρίνεται σε συστηματική και εντοπισμένη αμυλοείδωση και ταξινομείται ανάλογα με την ταυτότητα της πρωτεΐνης που σχηματίζει ινίδια. Μέχρι τώρα έχουν περιγραφεί >30 πρόδρομες πρωτεΐνες με αμυλοειδογόνο ιδιότητα. Όλες οι εναποθέσεις, εκτός από αδιάλυτα πρωτεϊνικά μόρια, περιέχουν επίσης γλυκοζαμινογλυκάνες και το αμυλοειδές P (SAP, serum amyloid P), μία γλυκοπρωτεΐνη που συνδέει το αμυλοειδές ανεξαρτήτως της πρωτεΐνης προέλευσης.
Οι συστηματικές αμυλοειδώσεις είναι νεοπλασματικής, φλεγμονώδους, γενετικής ή ιατρογενούς προέλευσης, ενώ οι τοπικές αμυλοειδώσεις έχουν σχέση μη τη γήρανση και το διαβήτη και εμφανίζονται σε μεμονωμένα όργανα χωρίς ένδειξη συστηματικής προσβολής.
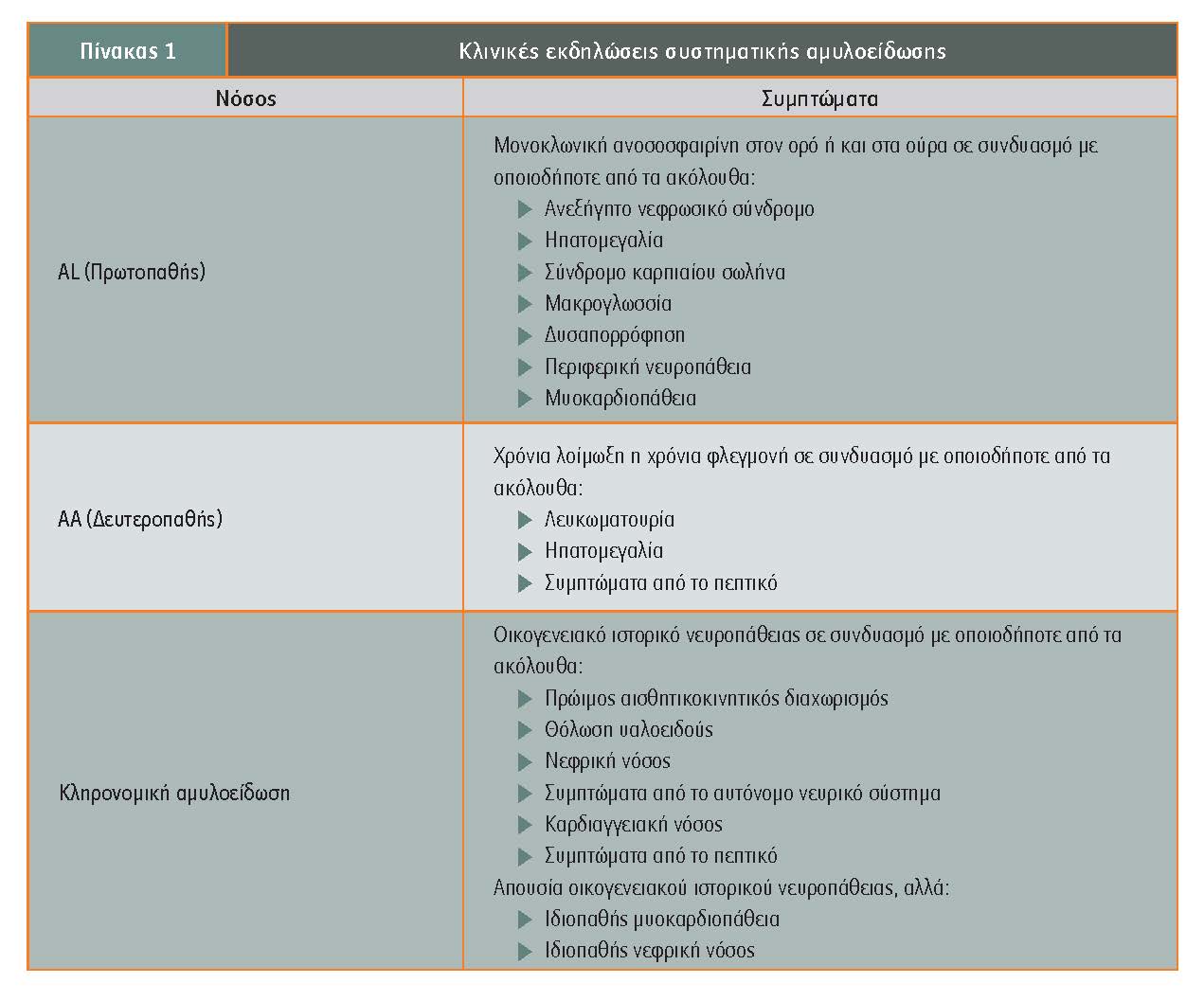
Η συχνότερη μορφή της συστηματικής αμυλοείδωσης είναι η πρωτοπαθής (AL) αμυλοείδωση ή αμυλοείδωση ελαφρών αλύσεων, προκαλούμενη από το σχηματισμό ινιδίων από θραύσματα ελαφρών αλύσεων (κ και λ) μονοκλωνικών αντισωμάτων. Σχετίζεται με πλασματοκυτταρικές δυσκρασίες (σε 20%-50% των ασθενών συνυπάρχει πολλαπλούν μυέλωμα).
Η δευτεροπαθής (AA) αμυλοείδωση εμφανίζεται συχνότερα ως επιπλοκή χρονίου φλεγμονώδους νοσήματος, όπως ρευματοειδής αρθρίτιδα, αγκυλοποιητική σπονδυλίτιδα, ιδιοπαθή φλεγμονώδη νοσήματα του εντέρου, λοιμώξεις όπως φυματίωση και οστεομυελίτιδα, αλλά και νεοπλάσματα.Οι ασθενείς που υποβάλλονται σε μακροχρόνια αιμοδιάλυση εναποθέτουν τη β2-μικροσφαιρίνη, παρουσιάζοντας αμυλοείδωση β2MG.Οι κλινικές εκδηλώσεις της νόσου είναι ποικίλες και εξαρτώνται αποκλειστικά από τη βιοχημική φύση των ινιδίων της πρωτεΐνης (πίνακας 1). Γενικά συμπτώματα περιλαμβάνουν την κόπωση, την ανορεξία και την απώλεια βάρους.
Εκδηλώσεις από το ήπαρ
Το ήπαρ συχνά προσβάλλεται από την αμυλοείδωση: 49%-95%, 70%-97% και 56%-60% συνολικά, σε ασθενείς με AL και AA αμυλοείδωση, αντίστοιχα. Την εξαίρεση αποτελεί η κληρονομική (οικογενής) αμυλοείδωση TTR, στην οποία το ήπαρ δε νοσεί. Παρά ταύτα, οι κλινικές εκδηλώσεις είναι συνήθως ήπιας έως μέτριας βαρύτητας και εμφανίζονται αργά στην πορεία της νόσου. Η συχνότερη εκδήλωση είναι η ηπατομεγαλία (57%-83%), η οποία δε συσχετίζεται με το βαθμό διήθησης του οργάνου.
Οι εναποθέσεις αμυλοειδούς στα πυλαία διαστήματα με συμπίεση των κολποειδών είναι δυνατόν να οδηγήσουν σε πυλαία υπέρταση μη κιρρωτικής αιτιολογίας, με δημιουργία κιρσών και ασκίτη. Σημειωτέον, ο ασκίτης συχνότερα οφείλεται σε καρδιακή ανεπάρκεια, υπολευκωματιναιμία και νεφρωσικό σύνδρομο παρά σε πυλαία υπέρταση. Για παράδειγμα, σε μια σειρά 47 ασθενών με αμυλοείδωση ήπατος μόνο ένας εμφάνισε ασκίτη χωρίς την παρουσία της πρωτεϊνουρίας.
Η συχνότερη εργαστηριακή ανωμαλία είναι η αύξηση της ALP (86%), περίπου 1/3 των ασθενών παρουσιάζουν τρανσαμινασαιμία. Η χολόσταση, η δυσλιπιδαιμία, οι διαταραχές πήξεως και η υπολευκωματιναιμία είναι οι πιο σπάνιες εκδηλώσεις. Τυπικό εύρημα (90% των ασθενών) είναι η ανεύρεση μονοκλωνικού κλάσματος ανοσοσφαιρινών στον ορό και ελεύθερων κ ή λ αλύσεως στον ορό ή και τα ούρα.
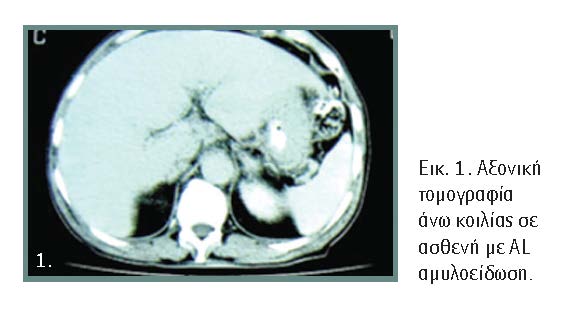
Οι απεικονιστικές εξετάσεις αναδεικνύουν την ηπατομεγαλία με αυξημένη ετερογένεια του παρεγχύματος (εικόνα 1), τα οποία σε συνδυασμό με αύξηση της ALP >1,5 φορές της τιμής αναφοράς αποτελούν έμμεσα στοιχεία προσβολής του οργάνου. Ο βαθμός σκληρότητας του ήπατος στην ελαστογραφία είναι αυξημένος λόγω διήθησης του οργάνου και όχι λόγω παρουσίας ίνωσης/κίρρωσης.
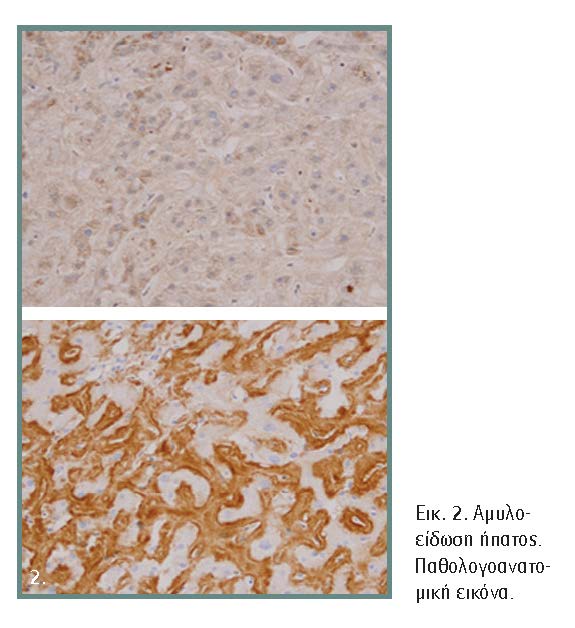
Η οριστική διάγνωση στηρίζεται στην ιστολογική εξέταση με ανεύρεση εναποθέσεων αμυλοειδούς, οι οποίες εμφανίζονται ροζ ή κόκκινες στο κοινό μικροσκόποιο και πράσινες στο πολωμένο φως ύστερα από ειδική χρώση με το ερυθρό του Congo (εικόνα 2). Οι δύο συχνότερες μορφές, AL και AA αμυλοείδωση, έχουν παρόμοια ιστολογικά ευρήματα και ο διαχωρισμός τους γίνεται με ανοσοϊστοχημική εξέταση.
Όσον αφορά τη βιοψία ήπατος σε αμυλοείδωση, πιστεύεται ότι ο κίνδυνος της αιμορραγίας ή και της ρήξης του ήπατος είναι ελαφρώς μεγαλύτερος συγκριτικά με ασθενείς με άλλα νοσήματα ήπατος. Συγκεκριμένα, στη βιβλιογραφία αναφέρονται μεμονωμένα περιστατικά αυτόματης ρήξης και της ενδοκοιλιακής αιμορραγίας σε ασθενείς με σημαντικού βαθμού διήθηση από αμυλοειδές, οι οποίες πιστεύεται ότι οφείλονται σε μηχανικά αίτια (αυξημένη εσωτερική πίεση του διηθημένου οργάνου σε ανελαστική κάψα του ήπατος).
Επίσης, οι ασθενείς συχνά παρουσιάζουν διαταραχές πήξεως, όπως η ανεπάρκεια του παράγοντα X, η ελάττωση των επιπέδων των παραγόντων πήξεων, εξαρτώμενων από τη βιταμίνη K, η αυξημένη δραστηριότητα της αντιθρομβίνης και της ινοδόλυσης, καθώς και την ενδοαγγειακή πήξη. Η διήθηση των τοιχωμάτων με ευθραυστότητα των αγγείων επίσης συμβάλλει σε αύξηση του κινδύνου αιμορραγίας. Επιπλέον, περιγράφονται περιστατικά αυτόματης ρήξης σπληνός, οφειλόμενα σε λειτουργικό υποσπληνισμό και διήθηση του οργάνου.
Η θεραπεία της αμυλοείδωσης αποσκοπεί στην καταστολή της παραγωγής των αδιάλυτων ινιδίων και διαφέρει αναλόγως του τύπου της αμυλοείδωσης. Στην πρωτοπαθή AL αμυλοείδωση η θεραπεία στοχεύει στην υποκείμενη πλασματοκυτταρική δυσκρασία, στη δευτεροπαθή αμυλοείδωση στην υποκείμενη φλεγμονή ή νεόπλασμα και στην τροποποίηση της μορφής της χρόνιας αιμοκάθαρσης ή μεταμόσχευση νεφρού στην αμυλοείδωση σχετιζόμενης με χρόνια νεφρική ανεπάρκεια.
Σε φάση κλινικών μελετών βρίσκονται φάρμακα τα οποία καταστρέφουν τις εναποθέσεις αμυλοειδούς σε όργανα κα ιστούς. Παράδειγμα αποτελεί η ουσία (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC που ελαττώνει τα επίπεδα SAP στον ορό και ακολουθείται από χορήγηση μονοκλωνικού αντισώματος IgG1 anti-SAP που διεγείρει την καταστροφή του SAP στις ιστικές εναποθέσεις.
Η μεταμόσχευση ήπατος έχει θέση σε κληρονομικές μορφές αμυλοείδωσης (π.χ. ΑΤΤΡm, AAPoAI, AFib) λόγω παραγωγής της πρόδρομων πρωτεϊνών από το ήπαρ.
Η πρόγνωση των ασθενών με αμυλοείδωση ήπατος είναι δυσμενής, ειδικά όταν υπάρχει χολόσταση. Η μέση επιβίωση χωρίς θεραπεία είναι 8-9 μήνες και με θεραπεία 10-14 μήνες. Η καρδιακή ανεπάρκεια και οι διαταραχές ρυθμού αποτελούν τις συχνότερες αιτίες θανάτου.
Bιβλιογραφία
Pinney JH, Hawkins PN. Amyloidosis. Ann Clin Biochem, 2012; 49: 229-241.
Nienhuis HL, Bijzet J, Hazenberg BP. The Prevalence and Managemen of Systemic Amyloidosis in Western Countries. Kidney Dis, 2016; 2: 10-19.
Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol., 2008; 103: 776-787.
Mousa AY, Abu-Halimah S, Alhalbouni S, et al. Amyloidosis and spontaneous hepatic bleeding, transcatheter therapy for hepatic parenchymal bleeding with massive intraperitoneal hemorrhage: a case report and review of the literature. Vascular, 2014; 22: 356-360.
Petre S, Shah IA, Gilani N. Review article: gastrointestinal amyloidosis – clinical features, diagnosis and therapy. Aliment Pharmacol Ther, 2008; 27: 1.006-1.016.
Νοέμβριος 2016
Διαβάστε επίσης: