








Γενετική και μοριακή διάγνωση κρανιοσκελετικών δυσπλασιών
Οι κρανιοσκελετικές δυσπλασίες αποτελούν ετερογενή ομάδα δυσπλασιών που επηρεάζουν το μέγεθος, το σχήμα και την οστεοποίηση (υφή και μετάλλωση) του σκελετού και του κρανιακού θόλου. Ανάλογα με το πρωτεύον χαρακτηριστικό των δυσπλασιών αυτών, μπορούν να διακριθούν σε 2 μεγάλες κατηγορίες: τις σκελετικές δυσπλασίες (ΣΔ), που επηρεάζουν κυρίως το σκελετό, και τις κρανιοσυνοστεώσεις (ΚΡ), που επηρεάζουν τον κρανιακό θόλο.
Γράφουν οι
Δέσποινα Αποστολοπούλου
Αγγελική Χατζάκη
Τμήμα Γενετικής και Μοριακής Βιολογίας, ΜΗΤΕΡΑ
Ολγα Καξίρα
Κανάρης Παναγόπουλος
Αλέξανδρος Στρατουδάκης
Ελληνικό Κρανιοπροσωπικό Κέντρο, ΜΗΤΕΡΑ
Σταύρος Σηφάκης
Μαιευτική και Γυναικολογική Κλινική
Πανεπιστημιακό Νοσοκομείο Ηρακλείου Κρήτης, Ηράκλειο
Στις κρανιοσκελετικές δυσπλασίες, ανάλογα με την κλινική οντότητα, επηρεάζονται:
• δύο είδη ιστών (οστό και χόνδρος),
• τρία είδη διαφοροποιημένων κυττάρων (οστεοβλάστες, οστεοκλάστες και χονδροκύτταρα),
• τα οργανικά συστατικά του οστού (π.χ. κολλαγόνο τύπου Ι),
• τα ανόργανα συστατικά του οστού (μετάλλωση) [π.χ. Ca10(PO4)6(OH)2].
Στις σκελετικές δυσπλασίες, τα επηρεαζόμενα μακρά οστά έχουν μεσοδερμική και εξωδερμική προέλευση (πρόδρομα κύτταρα: οστεοβλάστες, χονδροβλάστες, ινοβλάστες), ενώ στις κρανιοσυνοστεώσεις, τα επηρεαζόμενα κρανιακά οστά έχουν κυρίως εξωδερμική προέλευση, σχεδόν αποκλειστικά από κύτταρα της νευρικής ακρολοφίας (neural crest cells). Ο τρόπος διαφοροποίησης και ανάπτυξης του σκελετού και του κρανιακού θόλου διαφέρει.
Σε γενικές γραμμές, στο σκελετό υπερτερεί το πρότυπο ενδοχονδρικής οστεοποίησης (endochondral ossification), ενώ στον κρανιακό θόλο αυτό της διαμεμβρανικής οστεοποίησης (intramembranous ossification), και ως εκ τούτου σε κάθε κατηγορία δυσπλασιών βλάπτεται διαφορετικό αναπτυξιακό μονοπάτι και ενέχονται διαφορετικά αλλά και κοινά γονίδια (πίνακας 1).
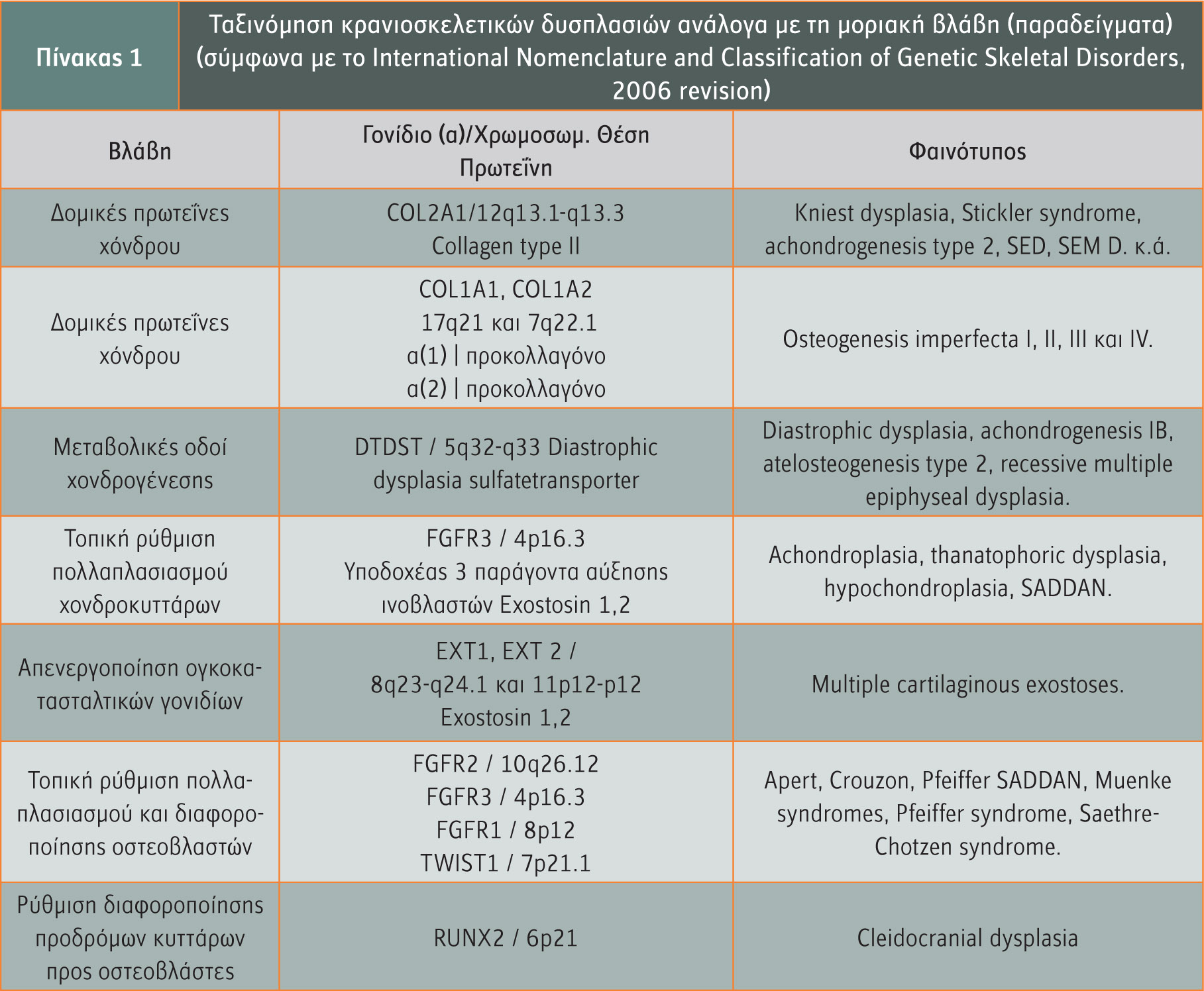
FGFRs-σχετιζόμενες σκελετικές δυσπλασίες
Συνολικά οι σκελετικές δυσπλασίες εμφανίζονται με συχνότητα 1/4.000 γεννήσεις, ενώ το 25% από τα έμβρυα με σκελετική δυσπλασία πεθαίνει ενδομητρίως και το 30% σε πρώιμη βρεφική ηλικία. Σημαντικός γενετικός παράγοντας που σχετίζεται άμεσα με τις κρανιοσκελετικές δυσπλασίες είναι οι υποδοχείς των αυξητικών παραγόντων των ινοβλαστών (FGFRs). Η οικογένεια των FGFRs, η οποία ανήκει στην κατηγορία των υποδοχέων κινάσης τυροσίνης, αποτελείται από 4 μέλη: FGFR1-4. Κάθε υποδοχέας αποτελείται βασικά από 3 εξωκυττάριες περιοχές που μοιάζουν με ανοσοσφαιρίνες (IGI-III), από μια διαμεμβρανική περιοχή (ΤΜ) και μια διαιρεμένη ενδοκυττάρια περιοχή κινάσης τυροσίνης (TK1/TK2) (σχήμα 1).
Η δημιουργία ενός τριμοριακού συμπλόκου, το οποίο περιλαμβάνει ένα μόριο αυξητικού παράγοντα των ινοβλαστών (FGF), που αποτελεί το μόριο-συνδέτη για τους FGFRs, ένα μόριο υποδοχέα (FGFR) και ένα μόριο πρωτεογλυκάνης (heparan sulfate proteoglycan) που βρίσκεται στην κυτταρική επιφάνεια, είναι απαραίτητη για το διμερισμό και την αυτοφωσφορυλίωση του υποδοχέα, με συνέπεια την ενεργοποίησή του. Αυτό έχει ως αποτέλεσμα την έναρξη μεταγωγής σήματος μέσω διαφόρων σηματοδοτικών μονοπατιών, που ελέγχουν κατά περίπτωση διάφορες σημαντικές κυτταρικές λειτουργίες, όπως ο πολλαπλασιασμός, η διαφοροποίηση, η μετανάστευση και η απόπτωση. Οι αυξητικοί παράγοντες των ινοβλαστών (FGFs) αποτελούν οικογένεια 22 δομικά παρόμοιων, μιτογόνων πολυπεπτιδίων και η σηματοδότηση που προάγουν, μέσω των υποδοχέων τους, παίζει πολύ κρίσιμο ρόλο στην πρώιμη εμβρυϊκή ανάπτυξη και στην οργανογένεση. Μεταλλαγές στα γονίδια που κωδικοποιούν για 3 από τους 4 υποδοχείς (FGFR1-3) έχουν καταγραφεί σε πληθώρα κρανιοσκελετικών ανωμαλιών.
Συγκεκριμένα, τρεις βασικές σκελετικές δυσπλασίες, η αχονδροπλασία (ACH), η υποχονδροπλασία (HCH) και η θανατηφόρος δυσπλασία τύπου 1 και 2 (TDI και TDII) συνδέονται με μεταλλαγές στο γονίδιο FGFR3.
Αχονδροπλασία
Η αχονδροπλασία (ACH) ή αχονδροπλασικός νανισμός (ΟΜΙΜ 100800) αντιπροσωπεύει περίπου το 15% του συνόλου των σκελετικών δυσπλασιών στη γέννηση και γι’ αυτό θεωρείται η συχνότερη μη θανατηφόρος σκελετική δυσπλασία, με ποσοστό που εκτιμάται από 1/15.000-1/25.000 γεννήσεις. Χαρακτηρίζεται από μικρό ανάστημα (ενήλικοι: άνδρες 131cm±5.6, γυναίκες 124cm±5.9), ριζομελική βράχυνση των μακρών οστών, έντονη οσφυϊκή λόρδωση, ελάττωση μεσοσπονδυλίων διαστημάτων στην περιοχή του οσφυοϊερού, βλαισο- και ραιβο- παραμορφώσεις κάτω άκρων, μακροκεφαλία και προέχον μέτωπο, υποπλασία οστών προσωπικού κρανίου και χαρακτηριστικό προσωπείο. Επιπλέον, συχνότατα εμφανίζονται περιορισμός της έκτασης και συστροφής του αγκώνα και σχήμα χεριών δίκην τρίαινας. Τα άτομα με αχονδροπλασία έχουν φυσιολογική νοητική ανάπτυξη και σπανιότατα εμφανίζουν ήπια νοητική υστέρηση και μαθησιακά προβλήματα.
Υποχονδροπλασία
Τα κλινικά χαρακτηριστικά της υποχονδροπλασίας (HCH) (OMIM 146000) είναι παρόμοια με αυτά της αχονδροπλασίας, αλλά ηπιότερα. Χαρακτηρίζεται από μικρό ανάστημα (128-165cm στους ενήλικες και 2-3 SD κάτω από το μέσο στα παιδιά), ριζομελική ή μεσομελική βράχυνση των μακρών οστών με ήπια πλάτυνση της μετάφυσης (μηριαίο και κνήμη) και βραχύ, πεπλατυσμένο αυχένα του μηριαίου. Επίσης, παρατηρούνται ελάττωση μεσοσπονδυλίων διαστημάτων στην κατώτερη οσφυϊκή μοίρα, γωνιώδη, βραχυμένα λαγόνια οστά και συχνά περιορισμός της έκτασης του αγκώνα, βραχυδακτυλία άνω και κάτω άκρων και γενικευμένη ήπια χαλαρότητα αρθρώσεων. Μακροκεφαλία συναντάται επίσης στα άτομα με υποχονδροπλασία, αλλά χωρίς υποπλασία οστών προσωπικού κρανίου και χαρακτηριστικό προσωπείο. Σπανιότερα, αλλά σημαντικά κλινικά χαρακτηριστικά είναι η σκολίωση, η οσφυϊκή λόρδωση, οι ήπιες βλαισο- και ραιβο- παραμορφώσεις κάτω άκρων, η οστεοαρθρίτιδα στους ενήλικες, η ήπια νοητική υστέρηση και τα μαθησιακά προβλήματα.=
Και στις δύο προαναφερθείσες δυσπλασίες ισχύουν τα εξής:
• Αυτοσωμική επικρατούσα κληρονομικότητα.
• Άνω του 85% των παιδιών με ACH από γονείς με φυσιολογικό ύψος (de novo μεταλλαγές).Τα υπόλοιπα από 1 ή 2 γονείς με ACH. 70% των παιδιών με HCH από γονείς με φυσιολογικό ύψος (de novo μεταλλαγές). Τα υπόλοιπα από 1 ή 2 γονείς με HCH.
• Ομόζυγη ACH θνησιγόνος.
• Οι de novo μεταλλαγές σχετίζονται με αυξημένη ηλικία του πατέρα.
• Όλες οι de novo μεταλλαγές κληρονομούνται από τον πατέρα και συμβαίνουν κατά τη σπερματογένεση, και πολύ σπανιότερα από μωσαϊκισμό (μόνο στην ACH).
• Στην ACH το χαμηλό ποσοστό γοναδικού μωσαϊκισμού εξηγεί την ελαφρά αυξημένη πιθανότητα γέννησης και 2ου παιδιού με ACH από το ίδιο ζευγάρι. Στην HCH δεν έχει αναφερθεί μωσαϊκισμός, άρα η πιθανότητα γέννησης και 2ου παιδιού με HCH από το ίδιο ζευγάρι είναι <0,01%.
• H διπλή ετεροζυγωτία για ACH και HCH είναι πολύ βαριά σκελετική δυσπλασία.
Θανατηφόρος δυσπλασία τύπου I και II
Η θανατηφόρος δυσπλασία (TDI: OMIM 187600, TDII: OMIM 187601) είναι η πιο συχνή σποραδική θνησιγόνος σκελετική δυσπλασία, με συχνότητα που κυμαίνεται μεταξύ 1/17.000-1/50.000 γεννήσεων. Τα κλινικά χαρακτηριστικά της περιλαμβάνουν μεγάλη βράχυνση των μακρών οστών, στενό θώρακα κωδωνοειδούς σχήματος αλλά φυσιολογικού μήκους, μακροκεφαλία με προέχον μέτωπο, χαμηλή πρόσφυση και επιπέδωση του ρινικού οστού, βραχυδακτυλία, υδροκέφαλο. Ειδικότερα, στον τύπο Ι παρατηρείται τοξοειδής κύρτωση του μηριαίου, το οποίο περιγράφεται σαν ακουστικό τηλεφώνου και ήπιος φαινότυπος κρανιοσυνοστέωσης, ενώ στον τύπο ΙΙ το μηριαίο είναι ευθύ, αλλά το κρανίο έχει το σχήμα τριφυλλιού λόγω πολλαπλής κρανιοσυνοστέωσης. Επίσης, μπορεί να βρεθούν υδρονέφρωση και καρδιακές ανωμαλίες. Τα πάσχοντα άτομα καταλήγουν λίγο μετά τη γέννηση λόγω αναπνευστικής ανεπάρκειας, η οποία οφείλεται σε υποπλασία των πνευμόνων. Κληρονομείται με τον αυτοσωμικό επικρατούντα χαρακτήρα και οφείλεται κυρίως σε de novo μεταλλαγές. Έχει επίσης αναφερθεί χαμηλό ποσοστό γοναδικού μωσαϊκισμού, στο οποίο οφείλεται η πιθανότητα επανεμφάνισης της δυσπλασίας στο ίδιο ζευγάρι, σε σπάνιες περιπτώσεις.
Κρανιοσυνοστεώσεις
Οι κρανιοσυνοστεώσεις είναι χωριστή ομάδα κρανιοσκελετικών δυσπλασιών με ποικίλη αιτιολογία, στις οποίες μόνο το 21% των περιπτώσεων οφείλεται σε γενετικά αίτια. Από αυτές, περίπου το 85% οφείλεται σε μεταλλαγές σε γονίδια, όπου κυρίαρχο ρόλο παίζουν και πάλι οι υποδοχείς των αυξητικών παραγόντων των ινοβλαστών (FGFRs 1-3), και το 15% σε χρωμοσωμικές ανωμαλίες. Άλλες αιτίες κρανιοσυνοστέωσης είναι ο ενδομήτριος περιορισμός της ανάπτυξης του εμβρύου και η έκθεση της εγκύου σε τερατογόνα, όπως το βαλπροϊκό και η ισοτρετινοΐνη.

Ο όρος κρανιοσυνοστέωση χρησιμοποιήθηκε για πρώτη φορά σε εγχειρίδιο παθολογικής ανατομίας το 1830 από τον Otto για να χαρακτηρίσει την πρόωρη σύντηξη των κρανιακών ραφών. Οι ραφές, οι στενές επιφάνειες επαφής των κρανιακών οστών, αποτελούνται από μη διαφοροποιημένο μεσέγχυμα και επιτρέπουν τη σύγχρονη με την ανάπτυξη του εγκεφάλου διαμόρφωση του κρανιακού θόλου. Η κρανιοσυνοστέωση εμφανίζεται με συχνότητα περίπου 1/2.500 γεννήσεις και μπορεί να διακριθεί σε μη συνδρομική και συνδρομική.
Οι μη συνδρομικές κρανιοσυνοστεώσεις, στις οποίες η πρόωρη σύντηξη των ραφών εμφανίζεται ως μεμονωμένο χαρακτηριστικό, μπορούν να ταξινομηθούν ανάλογα με τη ραφή η οποία συνοστεώνεται: μετωπιαία, οβελιαία, στεφανιαία και λαμβδοειδή. Ο φαινότυπος των κρανιοσυνοστεώσεων ποικίλλει ανάλογα με το ποια ή ποιες από τις ραφές θα συνοστεωθούν (π.χ. συνοστέωση της οβελιαίας ραφής προκαλεί σκαφοκεφαλία, των δύο στεφανιαίων βραχυκεφαλία, της μιας μόνο στεφανιαίας μετωπιαία πλαγιοκεφαλία, της μετωπιαίας και της οβελιαίας ακροκεφαλία, της μετωπιαίας τριγωνοκεφαλία, της λαμβδοειδούς και της οβελιαίας ινιακή πλαγιοκεφαλία και, τέλος, όλων των ραφών μικροκεφαλία). Η συχνότητα συνοστέωσης της κάθε ραφής έχει υπολογιστεί σε: 40%-55% στην οβελιαία, 20%-25% στη στεφανιαία (μονόπλευρη ή αμφοτερόπλευρη), 5%-15% στη μετωπιαία και 5% στη λαμβδοειδή. Στο 5%-15% των περιπτώσεων υπάρχει συνοστέωση σε περισσότερες από μία ραφές.
Στις συνδρομικές μορφές, η κρανιοσυνοστέωση μπορεί να αφορά μία ή περισσότερες ραφές και συνοδεύεται και από άλλα ευρήματα στα άκρα, στην καρδιά, στο κεντρικό νευρικό σύστημα κ.ά. Το 30% των συνδρόμων κρανιοσυνοστέωσης οφείλεται σε μεταλλαγές στα γονίδια FGFR1, FGFR2, FGFR3, TWIST1, EFNB1, MSX2 (Muscle segment homeobox) και RAB23 (Ras-associated protein). Επιπλέον, το 16% των συνδρομικών περιπτώσεων έχει συσχετιστεί, μέσω συμβατικών κυτταρογενετικών μεθόδων, με χρωμοσωμικές ανωμαλίες, κυρίως ελλείμματα και διπλασιασμούς, ενώ το ποσοστό αυτό αυξάνεται με τη χρήση σύγχρονων τεχνικών ανάλυσης των χρωμοσωμάτων (PMSA: polymorphic microsatellite segregation analysis, υποτελομεριδιακή MLPA: multiplex ligation-dependent probe amplification και array CGH: array-based comparative genomic hybridization), οι οποίες έχουν τη δυνατότητα να ανιχνεύουν μικροελλείμματα και μικροδιπλασιασμούς.
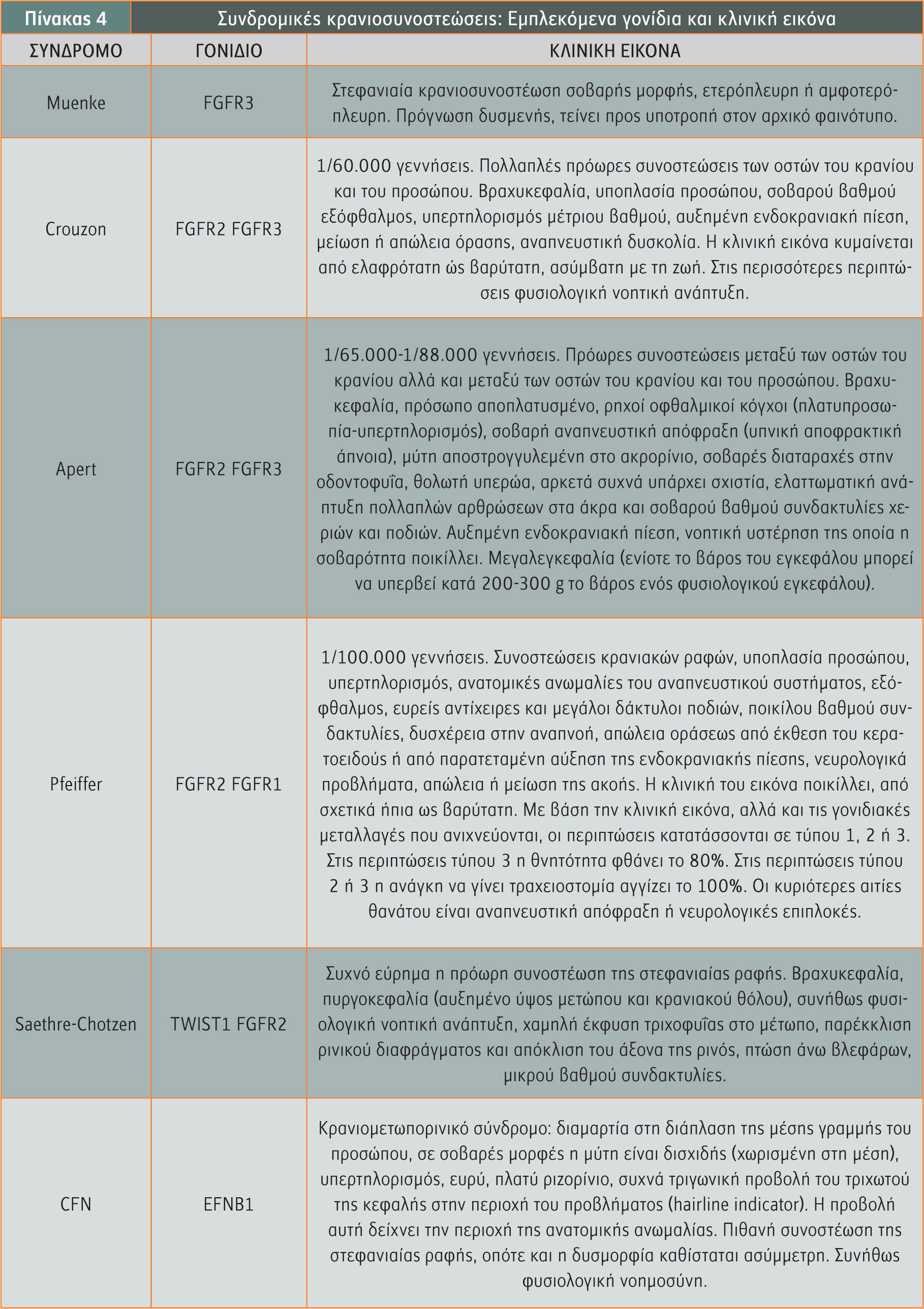
Τα συχνότερα σύνδρομα κρανιοσυνοστέωσης είναι τα: Muenke, Crouzon, Apert, Pfeiffer, Saethre-Chotzen και το σύνδρομο CFN (Craniofrontonasal) (σχήμα 2). Με εξαίρεση, ίσως, το σύνδρομο Apert, τα υπόλοιπα σύνδρομα κρανιοσυνοστέωσης παρουσιάζουν ετερογένεια τόσο στα κλινικά τους χαρακτηριστικά, ακόμα και μεταξύ μονοζυγωτικών διδύμων, όσο και στις μεταλλαγές στις οποίες οφείλονται. Το γεγονός αυτό καθιστά πολλές φορές δύσκολη την ακριβή διάγνωση, καθώς επίσης και την ανίχνευση του γενετικού αίτιου του συνδρόμου (πίνακας 4).
Μεθοδολογία μοριακής διάγνωσης
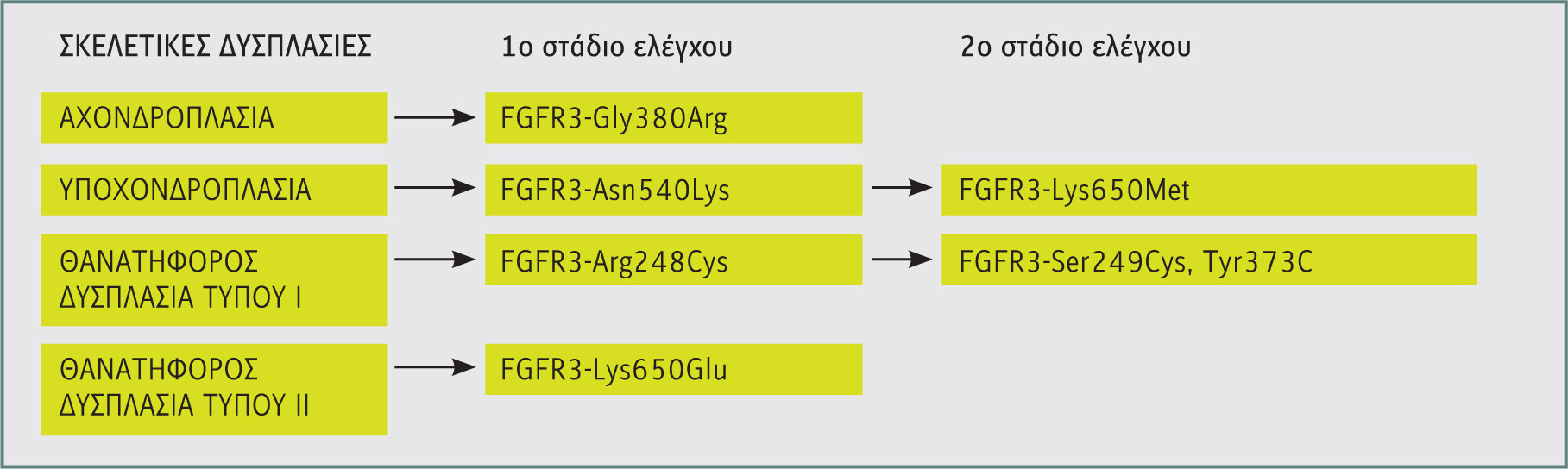
Η δυνατότητα εντοπισμού της γενετικής βάσης ποικίλλει ανάλογα με την κλινική εικόνα των κρανιοσκελετικών δυσπλασιών. Για τις σκελετικές δυσπλασίες και για ορισμένα σύνδρομα κρανιοσυνοστέωσης, η συσχέτιση κλινικού φαινοτύπου-γενετικής αιτιολογίας είναι άμεση, καθιστώντας έτσι πιο εύκολη τη μοριακή διάγνωση. Αντίθετα, στις υπόλοιπες συνδρομικές μορφές, καθώς και σε όλες τις μη συνδρομικές μορφές κρανιοσυνοστέωσης, ο συσχετισμός αυτός είναι πιο πολύπλοκος και η μοριακή διάγνωση πιο δύσκολη και χρονοβόρα.
Συγκεκριμένα, με τη διερεύνηση μιας και μόνο μεταλλαγής, διαφορετικής ανά περίπτωση σκελετικής δυσπλασίας, επιτυγχάνεται μοριακή διάγνωση για την αχονδροπλασία σε ποσοστό >98%, για την υποχονδροπλασία σε >70%, για τη θανατηφόρο δυσπλασία τύπου ΙΙ σε >99% και για το σύνδρομο Muenke στο 100%. Για τη διάγνωση του συνδρόμου Apert απαιτείται έλεγχος 2 μεταλλαγών που φθάνει σε ποσοστό 98%, ενώ για τη θανατηφόρο δυσπλασία τύπου Ι απαιτείται έλεγχος τριών διαφορετικών μεταλλαγών για να επιτευχθεί ποσοστό διάγνωσης 85%. Για τις υπόλοιπες περιπτώσεις, όπου η γενετική διάγνωση είναι πιο περίπλοκη, ακολουθείται ο αλγόριθμος που φαίνεται στους πίνακες 2 και 3.
Προγεννητική διάγνωση και γενετική συμβουλευτική
H προγεννητική διάγνωση των σκελετικών δυσπλασιών βασίζεται αρχικά στην 2-D υπερηχογραφία 2ου τριμήνου κατά τη 18η-23η εβδομάδα της κύησης, όπου απεικονίζεται η ανατομία του εμβρύου και υπολογίζονται λεπτομερώς τα βιομετρικά χαρακτηριστικά του σκελετού, του κρανίου και του θώρακα. Παρ? όλα αυτά, ο εντοπισμός υπερηχογραφικών δεικτών, διαγνωστικών για κάθε σκελετική ή κρανιακή δυσπλασία, σε έμβρυα χωρίς οικογενειακό ιστορικό είναι πολύ δυσχερής προσέγγιση. Κατά τα τελευταία χρόνια, η μοριακή διάγνωση έχει χρησιμοποιηθεί εκτενώς για την επίτευξη οριστικής διάγνωσης in utero. Σε αυτό το πλαίσιο, έχει γίνει εφικτή η προγεννητική διάγνωση των σκελετικών δυσπλασιών που οφείλονται στο γονίδιο FGFR3, με συνδυασμό δεδομένων που προκύπτουν από τη στενή συνεργασία του υπερηχογραφικού ελέγχου, του εργαστηρίου μοριακής διαγνωστικής και σε περιπτώσεις διακοπής της κύησης και του παθολογοανατομικού εργαστηρίου. Με βάση τον υπέρηχο 2ου τριμήνου από υπερηχογραφιστές ειδικευμένους στην ιατρική εμβρύου ως πρώτη διαγνωστική γραμμή, επιτυγχάνεται διαγνωστική ακρίβεια που κυμαίνεται από 31%-65% ανάλογα με τον τύπο της δυσπλασίας, ακρίβεια που τείνει να αυξάνεται στις πιο πρόσφατες μελέτες (67,9%). Όσον αφορά τις θανατηφόρες δυσπλασίες επιτυγχάνεται μεγαλύτερη ακρίβεια και πρωιμότερη διάγνωση, λόγω ειδικών υπερηχογραφικών δεικτών, ούτως ώστε να δίνεται η δυνατότητα έγκαιρης διακοπής της κύησης σε περίπτωση όπου επιλεγεί.
Άρα, ο ρόλος της υπερηχογραφίας εντοπίζεται στους παρακάτω στόχους:
• Να περιορίσει τη διαφορική διάγνωση σε συγκεκριμένα πιθανά γονίδια, ώστε να μπορεί να παρασχεθεί επιβεβαίωση ή αποκλεισμός με μοριακή προσέγγιση.
• Να προβλέψει τη θνησιμότητα in utero ή στην πρώιμη βρεφική ηλικία.
• Να εντοπίσει αρκετά πρώιμα την κρανιοσκελετική δυσπλασία, ώστε να παρασχεθεί έγκαιρα και αποτελεσματικά η διάγνωση και η γενετική συμβουλή.
Σε περίπτωση βέβαια, όπου υπάρχει οικογενειακό ιστορικό και ακριβής περιγραφή του φαινοτύπου του πάσχοντος ατόμου, η προγεννητική διάγνωση είναι σαφώς ευκολότερη, αν και στην πλειονότητα των περιπτώσεων, δεδομένου ότι οι μεταλλάξεις εμφανίζονται de novo, τα ευρήματα πιθανόν να περιορίζονται στον τυχαίο εντοπισμό βράχυνσης των μακρών οστών ή σε ανώμαλο σχήμα του κρανιακού θόλου.
Νοέμβριος 2012














