








Γενετική του καρκίνου
Περίπου το 10% όλων των μορφών καρκίνου οφείλονται στη βλάβη ενός γονιδίου, η οποία έχει κληρονομηθεί. Από τις πιο συχνές μορφές καρκίνου που έχουν κληρονομική προδιάθεση είναι ο οικογενής καρκίνος του μαστού και των ωοθηκών, η οικογενής πολυποδίαση του παχέος εντέρου και ο οικογενής χωρίς πολυποδίαση καρκίνος του παχέος εντέρου ή σύνδρομο Lynch.
Γράφουν οι
Λίνα Φλωρεντίν
Μοριακή Βιολόγος – Κυτταρογενετίστρια
Διευθύντρια ΑλφαLab – Κέντρο Μοριακής Βιολογίας και Κυτταρογενετικής
Γεώργιος Δ. Λύπας
Παθολόγος Ογκολόγος
Υπεύθυνος Μονάδας Γενετικής Ογκολογίας ΥΓΕΙΑ
Και στα τρία αυτά νοσήματα η κληρονόμηση γίνεται με αυτοσωμικό επικρατή χαρακτήρα, δηλαδή ο φορέας μιας μετάλλαξης έχει μεγάλη πιθανότητα να νοσήσει και 50% πιθανότητα να μεταφέρει τη βλάβη στην επόμενη γενιά. Κάθε άνθρωπος μπορεί να κληρονομήσει μια μετάλλαξη τόσο από τη μητέρα του όσο και από τον πατέρα του. Γι’ αυτόν το λόγο πρέπει στο οικογενειακό ιστορικό του ατόμου που προσέρχεται για έλεγχο μεταλλάξεων να καταγράφονται πληροφορίες για τους συγγενείς μητρικής και πατρικής προέλευσης. Από τα παραπάνω σαφώς προκύπτει ότι, προκειμένου να επιλεγεί το κατάλληλο γονίδιο προς μελέτη και να γίνει σωστή ανάλυση και ερμηνεία των αποτελεσμάτων, είναι απαραίτητη η καταγραφή όσο το δυνατόν πληρέστερου οικογενειακού ιστορικού πασχόντων και μη, σε τρεις ή τέσσερις γενεές, στο οποίο θα φαίνονται η ηλικία εμφάνισης της νόσου και όλες οι άλλες μορφές καρκίνου που μπορεί να έχουν διαγνωστεί στην οικογένεια. Σήμερα είναι γνωστό και πρέπει να λαμβάνεται υπόψη ότι η εμφάνιση του καρκίνου είναι αποτέλεσμα πολλών γενετικών και περιβαλλοντικών παραγόντων.
Η σωστή αντιμετώπιση τέτοιων περιπτώσεων πρέπει να είναι το αποτέλεσμα στενής συνεργασίας του θεράποντος ογκολόγου, της μονάδας γενετικής ογκολογίας και του εργαστηρίου γενετικής.
Κληρονομικός καρκίνος του μαστού και των ωοθηκών
Το 5%-10% του συνόλου των καρκίνων του μαστού ή και των ωοθηκών έχουν κληρονομική προδιάθεση. Ο κληρονομικός καρκίνος του μαστού, αλλά και των ωοθηκών οφείλεται κατά κύριο λόγο σε μεταλλάξεις των γονιδίων BRCA1 και BRCA2. Συναντώνται, όμως, και γενετικά νοσήματα προδιάθεσης ανάπτυξης καρκίνου του μαστού, τα οποία συνδέονται με άλλα γονίδια, όπως τα TP53, PTEN, ATM και CDH1. Παραδείγματα αυτών είναι η νόσος του Cowden, τα σύνδρομα Peutz-Jeghers και Li Fraumeni, η αταξία-τηλαγγειεκτασία κ.ά. Επίσης υφίστανται νοσήματα προδιάθεσης καρκίνου των ωοθηκών, όπως το σύνδρομο Gorlin, ο οικογενής χωρίς πολυποδίαση καρκίνος του παχέος εντέρου κ.ά. Αποκαλύφθηκαν επίσης γονίδια ενδιαμέσου διεισδυτικότητας, όπως το CHEK2, και πολυμορφισμοί (SNPs) που θεωρούνται γενετικοί προδιαθεσιακοί παράγοντες χαμηλής διεισδυτικότητας, οι οποίοι μπορούν να τροποποιήσουν τον κίνδυνο εμφάνισης καρκίνου του μαστού σε μια ή ένα φορέα όπως o πολυμορφισμός SNP 135G>C RAD51.
Το γονίδιο BRCA1 βρίσκεται στο χρωμόσωμα 17 και αποτελείται από 24 εξώνια, ενώ το γονίδιο BRCA2 βρίσκεται στο χρωμόσωμα 13 και αποτελείται από 27 εξώνια. Περίπου το 40% των κληρονομικών περιπτώσεων καρκίνου μαστού οφείλεται σε αλλαγές στα παραπάνω γονίδια. Μεταλλάξεις στο γονίδιο BRCA1 εντοπίζονται στο 45% των οικογενειών με πολλές περιπτώσεις καρκίνου μαστού και στο 90% των οικογενειών με καρκίνο μαστού και ωοθηκών. Μεταλλάξεις στο γονίδιο BRCA2 εντοπίζονται στο 35% των οικογενειών με πολλές περιπτώσεις καρκίνου μαστού, καρκίνο μαστού σε άρρενα άτομα, καρκίνο ωοθηκών και προστάτη, παγκρέατος.
Τα κριτήρια επιλογής των δειγμάτων για ανάλυση των BRCA1 και BRCA2 γονιδίων είναι τα παρακάτω:
Για ασθενείς που έχουν νοσήσει με καρκίνο, όταν συντρέχουν ένα ή περισσότερα από τα εξής:
– Διάγνωση καρκίνου του μαστού σε σχετικά νέα ηλικία (πριν από τα 50).
– Τριπλά αρνητικός καρκίνος μαστού (ER-, PR-, HER2-).
– Δύο πρωτοπαθείς καρκίνοι μαστού σε ένα άτομο.
– Καρκίνος μαστού σε οποιαδήποτε ηλικία και επιπλέον:
• Ένας ή περισσότεροι συγγενείς εξ αίματος με καρκίνο μαστού πριν τα 50,
• Ένας ή περισσότεροι συγγενείς εξ αίματος με καρκίνο ωοθήκης ή σαλπίγγων ή πρωτοπαθή καρκίνο περιτοναίου, ανεξαρτήτως ηλικίας διάγνωσης,
• Δύο ή περισσότεροι συγγενείς εξ αίματος με καρκίνο του μαστού ή/και καρκίνο παγκρέατος σε οποιαδήποτε ηλικία.
– Καρκίνος των ωοθηκών/σαλπίγγων/πρωτοπαθής καρκίνος περιτοναίου.
– Καρκίνος του μαστού σε άνδρα.
Για υγιή άτομα:
– Δύο ή περισσότερα άτομα με καρκίνο του μαστού στην ίδια πλευρά της οικογένειας.
– Ένα ή περισσότερα άτομα με καρκίνο της ωοθήκης.
– Συγγενής εξ αίματος με γνωστή μετάλλαξη στα γονίδια BRCA1/2.
– Συγγενής εξ αίματος, άνδρας, με καρκίνο μαστού.
– Καταγωγή από ομάδα υψηλού κινδύνου (Eβραίοι Ashkenazi).
Κληρονομική πολυποδίαση του παχέος εντέρου
Η οικογενής πολυποδίαση (Familial Adenomatous Polyposis – FAP) χαρακτηρίζεται από την παρουσία εκατοντάδων έως και χιλιάδων πολυπόδων στο παχύ έντερο, αλλά και στο ορθό, κατά τη δεύτερη δεκαετία της ζωής του ατόμου. Σχεδόν όλοι οι ασθενείς θα αναπτύξουν καρκίνο του παχέος εντέρου αν δε διαγνωστούν εγκαίρως και δεν ακολουθήσουν την κατάλληλη θεραπεία σε πρώιμο στάδιο. Το 15% του συνόλου των περιπτώσεων καρκίνου του παχέος εντέρου είναι κληρονομούμενο, με την οικογενή πολυποδίαση του παχέος εντέρου να ευθύνεται για το 1% περίπου των περιπτώσεων.
Η ηπιότερη μορφή της πάθησης (attenuated FAP – AFAP) χαρακτηρίζεται από την παρουσία πολύ μικρότερου αριθμού πολυπόδων (10-100). Οι πολύποδες αναπτύσσονται κατά την τρίτη δεκαετία της ζωής του ατόμου, ενώ ο ασθενής εμφανίζει καρκίνο στην τέταρτη ή την πέμπτη δεκαετία της ζωής του. Η πλειονότητα των FAP ασθενών έχει βεβαρημένο οικογενειακό ιστορικό με πολύποδες και καρκίνο του παχέος εντέρου.
Η οικογενής πολυποδίαση οφείλεται συνήθως σε μεταλλάξεις του ογκοκατασταλτικού γονιδίου APC, το οποίο βρίσκεται στο χρωμόσωμα 5 και αποτελείται από 15 εξώνια. Τα άτομα που έχουν κληρονομήσει μια μετάλλαξη στο γονίδιο APC έχουν 100% πιθανότητα να εμφανίσουν κακοήθεια, όμως οι πιθανότητες αυτές μειώνονται σημαντικά αν υποβληθούν σε έγκαιρη αποκάλυψη της νόσου, γίνει δηλαδή προσυμπτωματικός έλεγχος και επιλεγεί θεραπευτική προσέγγιση (βλέπε συνέχεια).
Ένα 25% περίπου φέρει μεταλλάξεις στο γονίδιο ΜUTYH στο χρωμόσωμα 1 (15 εξώνια). Η νόσος κληρονομείται με αυτοσωμικό υπολειπόμενο χαρακτήρα, δηλαδή χρειάζονται 2 αντίγραφα του μεταλλαγμένου γονιδίου για να νοσήσει ένα άτομο και οι γονείς του είναι ασυμπτωματικοί φορείς της νόσου. Οι ασθενείς με μεταλλάξεις στο ΜUTYH έχουν κλινική εικόνα παρόμοια με την ηπιότερη μορφή της οικογενούς πολυποδίασης.
Τα κριτήρια επιλογής των ατόμων για ανάλυση των APC και ΜUTYH γονιδίων είναι τα παρακάτω:
– Ιστορικό καρκίνου του παχέος εντέρου και ορθού, σε συγγενείς εξ αίματος και σε ηλικία – Ιστορικό με πολύποδες στο έντερο σε ηλικία – Οικογενειακό ιστορικό με ελκώδη κολίτιδα.
– Εάν έχουν παρατηρηθεί στο υπό εξέταση άτομο συμπτώματα όπως εύκολη κόπωση, αδυναμία, αλλαγή στις συνήθειες του εντέρου, διάρροια ή δυσκοιλιότητα, ερυθρό ή σκούρο αίμα στα κόπρανα, απώλεια βάρους, κοιλιακά άλγη, κωλικοί, φούσκωμα.
– Εάν υπάρχει στην οικογένεια ιστορικό με καρκίνο του μαστού ή της μήτρας.
Κληρονομικός χωρίς πολυποδίαση καρκίνος του παχέος εντέρου (HNPCC) ή σύνδρομο Lynch
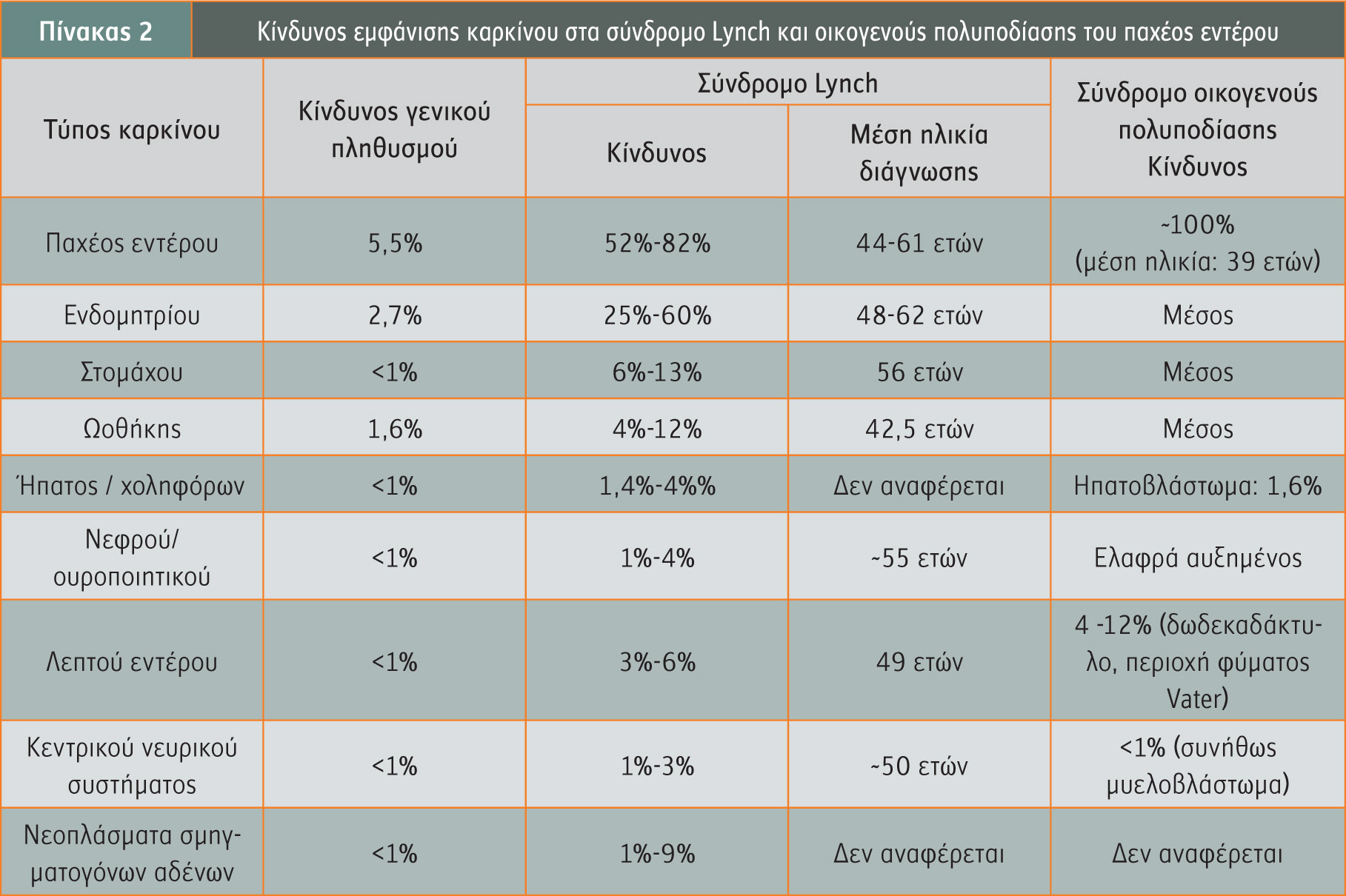
Στις οικογένειες με σύνδρομο Lynch εντοπίζονται πάσχοντες σε όλες τις γενιές, και σε νεαρή ηλικία. Ορισμένοι απ’ αυτούς τους ασθενείς έχουν αυξημένο κίνδυνο να αναπτύξουν και άλλες μορφές καρκίνου (βλέπε πίνακα 2). Η συχνότητα εμφάνισης του συνδρόμου Lynch είναι 1%-2% των περιπτώσεων καρκίνου του παχέος εντέρου πριν από την ηλικία των 60 ετών.
Οι όγκοι που αναπτύσσονται σε άτομα οικογενειών με HNPCC προκαλούνται κατά κύριο λόγο από μεταλλάξεις στα γονίδια MLH1, MSH2, MSH6 υπεύθυνα για την επιδιόρθωση στο DNA (MisMatch Repair genes – MMR). Μεταλλάξεις σε αυτά τα γονίδια αναλογούν στο 90% περίπου των οικογενειών (60%, 30%, 7%-10% αντιστοίχως) που έχουν κλινικά διαγνωστεί με σύνδρομο Lynch. Σπανιότερα ανιχνεύονται μεταλλάξεις στα PSM2 και EPCAM γονίδια.
Οι μεταλλάξεις σε αυτά τα γονίδια δίνουν χαρακτηριστική εικόνα στα καρκινικά κύτταρα που ονομάζεται μικροδορυφορική αστάθεια (MicroSatellite Instability-MSI). Ο έλεγχος για MSI γίνεται σύμφωνα με τα κριτήρια της Bethesda και είναι σημαντικό κριτήριο βάσει του οποίου επιλέγεται ο έλεγχος μεταλλάξεων στα MMR γονίδια.
Για τη γενετική διερεύνηση ατόμων ύποπτων για σύνδρομο Lynch είναι τα γνωστά κριτήρια της Bethesda και τα κριτήρια του Amsterdam II. Τα μεν έχουν μεγαλύτερη ευαισθησία, τα δε μεγαλύτερη ειδικότητα. Σύμφωνα με αυτά:
– Πρέπει να υπάρχουν τουλάχιστον τρεις ασθενείς στη ίδια οικογένεια με διαγνωσμένο καρκίνο των οποίων η ιστολογική εξέταση τους κατατάσσει στους καρκίνους που συνδέονται με το σύνδρομο Lynch.
– Πρέπει να υπάρχουν περιπτώσεις καρκίνου του παχέος εντέρου ανάμεσα σε συγγενείς 1ου βαθμού σε δυο τουλάχιστον γενεές.
– Πρέπει σε τουλάχιστον μία από τις περιπτώσεις αυτές η διάγνωση να έχει γίνει πριν από την ηλικία των 50 ετών.
– Να έχει χαρακτηριστεί ο όγκος, έπειτα από βιοψία, με έλεγχο μικροδορυφορικής αστάθειας MSI.
Πρόσφατα αναπτύχθηκε το μαθηματικό μοντέλο ΙPREMM 1,2,6» για τον αριθμητικό υπολογισμό πιθανοτήτων ύπαρξης συνδρόμου Lynch, του οποίου η ευαισθησία υπερέχει και των κριτηρίων της Bethesda, το οποίο τροφοδοτείται με δεδομένα οικογενειακού ιστορικού καρκίνου συγγενών 1ου και 2ου βαθμού (βλ. σύνδεσμο http://dana farber.prod.dfcidev.org/pat/cancer/gastrointestinal/crc-calculator/default.asp). Τα άτομα των οποίων οι πιθανότητες είναι ίσες ή μεγαλύτερες του 5%, χρήζουν γενετικής αξιολόγησης. Εντούτοις, στα μεγάλα νοσοκομεία των ΗΠΑ κερδίζει διαρκώς έδαφος η τάση για την εφαρμογή διαγνωστικού αλγορίθμου σε όλους τους ασθενείς με καρκίνο του παχέος εντέρου και του ενδομητρίου. Συγκεκριμένα, καθιερώθηκε ως έλεγχος ρουτίνας η ανίχνευση δεικτών μικροδορυφορικής αστάθειας (MSI) και η ανοσοϊστοχημική μελέτη του όγκου για έκφραση των πρωτεϊνών της οικογένειας MMR (MLH1, MSH2, MSH6 & PMS2). Η απουσία έκφρασης μιας εκ των πρωτεϊνων αυτών ή/και η μικροδορυφορική αστάθεια του όγκου, επιβάλλουν τη γενετική αξιολόγηση του ασθενούς. Εξ αυτών, 1 στους 3 θα αποδειχθεί ότι φέρει το σύνδρομο Lynch.
Με τον τρόπο αυτόν η ανίχνευση των φορέων του συνδρόμου Lynch αγγίζει το 100%. Επιπλέον, σχετικές μελέτες κόστους-αποτελεσματικότητας έχουν δείξει την υπεροχή αυτής της στρατηγικής σε επίπεδο πληθυσμού και ταυτόχρονα τη δυνατότητα μείωσης της νοσηρότητας από καρκίνο του παχέος εντέρου κατά 13,4% και του καρκίνου του ενδομητρίου κατά 8,5%.
Μεθοδολογίες ανίχνευσης μεταλλάξεων και ερμηνεία αποτελεσμάτων-Μοριακή ανάλυση μικροδορυφορικής αστάθειας (MSI)
Το 70% -85% των βλαβών που εντοπίζονται στα γονίδια που προαναφέρθηκαν, είναι σημειακές μεταλλάξεις ενώ στο υπόλοιπο ποσοστό εντοπίζονται μεγάλες ελλείψεις ή διπλασιασμοί μέρους των γονιδίων. Μεταλλάξεις είναι δυνατό να ανιχνευθούν σε όλο το μήκος των γονιδίων και για το λόγο αυτό είναι απαραίτητος ο έλεγχος όλων των εξωνίων και όχι μόνο μέρους τους.
Υπάρχουν δύο ειδών τεχνικές για την ανίχνευση μεταλλάξεων και στις τρεις περιπτώσεις κληρονομικού καρκίνου που περιγράφονται παραπάνω:
– Καθορισμός της πρωτοταγούς δομής του DNA (Direct Sequencing) χρησιμοποιείται για την ανίχνευση σημειακών μεταλλάξεων.
– Multiplex Ligation-dependent Probe Amplification (MLPA) χρησιμοποιείται για την ανίχνευση μεγάλων ελλείψεων και μεγάλων διπλασιασμών.
Συνήθως γίνεται αρχικά o καθορισμός της πρωτοταγούς δομής του DNA και, επί αρνητικού αποτελέσματος, γίνεται έλεγχος ελλείψεων και διπλασιασμών με MLPA.
Σημειώνεται ότι ο έλεγχος της μικροδορυφορικής αστάθειας MSI γίνεται με συγκριτική ανάλυση συγκεκριμένων πολυμορφικών δεικτών σύμφωνα με τα κριτήρια της Bethesda ανάμεσα στο DNA που απομονώνεται από τον όγκο και το DNA που απομονώνεται από το περιφερικό αίμα του ασθενούς.
Κατά τη διάρκεια της ανάλυσης υπάρχει περίπτωση να ανιχνευθούν:
1. Γνωστές και καλά χαρακτηρισμένες μεταλλάξεις, οι οποίες συνδέονται με τη νόσο ή νέες μεταλλάξεις που η θέση τους και το είδος τους με σαφήνεια οδηγούν σε δυσλειτουργία του γονιδίου και είναι η αιτία της νόσου (παθολογική μετάλλαξη).
2. Πολυμορφισμοί, οι οποίοι θεωρούνται φυσιολογικές παραλλαγές καθώς απαντούνταν και στο γενικό πληθυσμό και δεν έχουν καμία κλινική σημασία.
3. Ακατηγοριοποίητες παραλλαγές των οποίων η θέση και το είδος δεν είναι σαφές εάν προκαλούν δυσλειτουργία του γονιδίου και για το λόγο αυτό θεωρούνται παραλλαγές αγνώστου κλινικής σημασίας (mutations of clinical insignificance) και στην περίπτωση αυτήν η μελέτη άλλων υγιών και πασχόντων ατόμων της οικογένειας μπορεί να βοηθήσει στην αποσαφήνιση της κλινικής σημασίας της παραλλαγής αυτής.
Για την επιλογή της κατάλληλης εργαστηριακής διαγνωστικής προσέγγισης και της σωστής ερμηνείας των αποτελεσμάτων από τον εργαστηριακό γενετιστή είναι απαραίτητο να παρέχονται από τον κλινικό ιατρό ή από τον κλινικό γενετιστή όλες οι πληροφορίες του οικογενειακού ιστορικού του ασθενούς. Όπου αυτό είναι εφικτό, είναι προτιμότερο ο μοριακός έλεγχος για την ύπαρξη ή όχι κάποιας μετάλλαξης στα γονίδια αυτά να γίνεται αρχικά σε πάσχον μέλος της οικογένειας. Στη συνέχεια, εφόσον ανιχνευθεί βλάβη, μπορεί να γίνει μοριακός έλεγχος σε όποια από τα υγιή μέλη της οικογένειας το επιθυμούν (προσυμπτωματικός έλεγχος) ώστε να διαπιστωθεί εάν είναι φορείς της μετάλλαξης. Στην περίπτωση όπου δεν είναι εφικτός ο έλεγχος σε πάσχον μέλος της οικογένειας, η μη ανίχνευση μετάλλαξης σε κάποιο από τα γονίδια σε υγιές μέλος της οικογένειας δεν συνεπάγεται πάντα απουσία κληρονομούμενης μετάλλαξης στην οικογένεια.
Πρόσφατα άρχισε να εφαρμόζεται σε ειδικές περιπτώσεις μια νέας γενιάς μέθοδος, η οποία ονομάζεται next generation sequencing και είναι μια μέθοδος ανίχνευσης μεταλλάξεων νέας γενιάς, με την οποία είναι δυνατή η ανίχνευση πολλών γονιδίων συγχρόνως. Αναπτύσσονται λοιπόν νέα πρωτόκολλα ανίχνευσης ομάδων γονιδίων όπως αυτά για τους γυναικολογικούς καρκίνους, όπου 18 γονίδια εκτός των BRCA1/2 μελετώνται (ΑTM, BARD1, BRIP1, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2,MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, STK11 και TP53) ή για τον καρκίνο του παχέος εντέρου με 14 γονίδια (APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2,MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11 και TP53). Ο προγεννητικός έλεγχος, καθώς και η προεμφυτευτική διάγνωση (PGD) είναι εφικτά εάν είναι γνωστή η μετάλλαξη της οικογενείας, παρ’ όλα αυτά, λόγω του ότι πρόκειται σε ορισμένες περιπτώσεις, για νόσηματα που εμφανίζονται σε μεγαλύτερη ηλικία (εφηβεία κ.λπ.) η οικογένεια σκόπιμο είναι να δεχθεί γενετική συμβουλή πριν ληφθούν σημαντικές αποφάσεις για την απόκτηση παιδιού που δεν θα πάσχει.
Στρατηγικές μείωσης κινδύνου/πρόληψης σε οικογένειες με κληρονομική προδιάθεση]
Η διαδικασία της γενετικής αξιολόγησης και του γονιδιακού ελέγχου προσφέρει πολύ περισσότερα από την απάντηση του ερωτήματος που απασχολεί συχνά τους καρκινοπαθείς: γιατί σε μένα; Σήμερα είναι τεκμηριωμένη η αποτελεσματικότητα διαφόρων στρατηγικών μείωσης κινδύνου και πρόληψης των συχνότερων μορφών κληρονομικού καρκίνου. Ενδεικτικά αναφέρονται κάποιες κύριες επιλογές οι οποίες συνοψίζονται πιο κάτω με βάση τις σχετικές συστάσεις των διεθνών επιστημονικών εταιρειών (όπως National Comprehensive Cancer Network των ΗΠΑ, European Society of Medical Oncology κ.τ.λ.).
Φορείς παθογόνων μεταλλάξεων γονιδίου BRCA1/2:
– Έναρξη προσυμπτωματικού ελέγχου (screening) των μαστών, με μαστογραφία και μαγνητική μαστογραφία από τα 25 έτη (ή 10 χρόνια νωρίτερα από το πρωιμότερο κρούσμα στην οικογένεια).
– Χημειοπροφύλαξη, δηλαδή προφυλακτική λήψη ταμοξιφένης επί 5 έτη.
– Προφυλακτική αμφοτερόπλευρη μαστεκτομή (με ή χωρίς τη διατήρηση του δέρματος). Μείωση του κινδύνου για καρκίνο του μαστού κατά 90%-95%.
– Προφυλακτική αμφοτερόπλευρη σαλπιγγοωοθηκεκτομή, για πρόληψη καρκίνου ωοθηκών/σαλπίγγων (σε ηλικια 35-38 ετών και εφόσον έχει ολοκληρώσει η γυναίκα τα σχέδια τεκνοποίησης).
Προσυμπτωματικός έλεγχος για καρκίνο προστάτη, παγκρέατος ή/και κακόηθες μελάνωμα, βασίζονται στο λοιπό οικογενειακό ιστορικό.
Φορείς οικογενούς πολυποδιάσης του παχέος εντέρου:
– Για την κλασική μορφή του FAP (πολυποδίαση σε παιδική ηλικία) συνιστάται προφυλακτική κολεκτομή από την ηλικία περίπου των 12 ετών, καθώς ο κίνδυνος ΚΠΕ ως την ηλικία των 45 ετών αγγίζει το 100%.
– Επίσης, για την πρόληψη εξωεντερικών νεοπλασμάτων συνιστάται τακτικός έλεγχος του ανωτέρου πεπτικού (έλεγχος στομάχου και δωδεκαδακτύλου) από την ηλικία των 20 περίπου ετών και επανάληψη ανά 1-3 έτη, με βάση τα ευρήματα.
– Στην περίπτωση ήπιας μορφής του συνδρόμου (aFAP), συνιστάται ετήσιος ενδοσκοπικός έλεγχος του κατωτέρου και ανωτέρου πεπτικού. Αν ευρίσκεται μεγάλος αριθμός πολυπόδων, είναι εύλογη και η συζήτηση για το ενδεχόμενο της προφυλακτικής κολεκτομής.
Φορείς συνδρόμου Lynch
– Για υγιείς φορείς: έναρξη προσυμπτωματικού ελέγχου με κολονοσκόπηση σε ηλικία 25 ετών ή 10 έτη πριν από το πρωιμότερο κρούσμα καρκίνου στην οικογένεια.
– Για νεοδιαγνωσθέντες ασθενείς με εξαιρέσιμο ΚΠΕ και δεδομένου ότι ο κίνδυνος για νέο πρωτοπαθή ΚΠΕ είναι υψηλός, πρέπει να εξετάζεται σοβαρά και η επιλογή της υφολικής κολεκτομής. Σε αυτούς τους ασθενείς και εντός δεκαετίας από την αρχική διάγνωση, μόλις το 3% αυτών που επιλέγουν υφολική κολεκτομή εμφανίζει νέο ΚΠΕ έναντι 16% των ασθενών που επιλέγουν εκτομή του πάσχοντος τμήματος.
– Ενδοσκοπικός έλεγχος ανώτερου πεπτικού κατά τη διάγνωση του συνδρόμου και εν συνεχεία επανάληψη με βάση τα ευρήματα.
– Έλεγχος του ουροποιητικού (με απεικόνιση και κυτταρολογική εξέταση ούρων).
– Απεικονιστικός έλεγχος παγκρέατος, εφόσον υπάρχει σχετικό ιστορικό.
– Οι γυναίκες θα πρέπει επιπροσθέτως να κάνουν ετήσιο γυναικολογικό έλεγχο.

Νοέμβριος 2012














